Julien Allali
MiGaL Tutorial
MiGaL Tutorial: Example 2, building phylogenetic trees
This example shows how to compute the scoring matrix for a set of structures and to build a phylogenetic tree with the phylip neighbor joining method.
The data
For this example, we take 6 16S rRNA from The Comparative RNA Web Site.
You can download these structures here.
Uncompress the data in a directory using tar -zxf 16S.tgz.
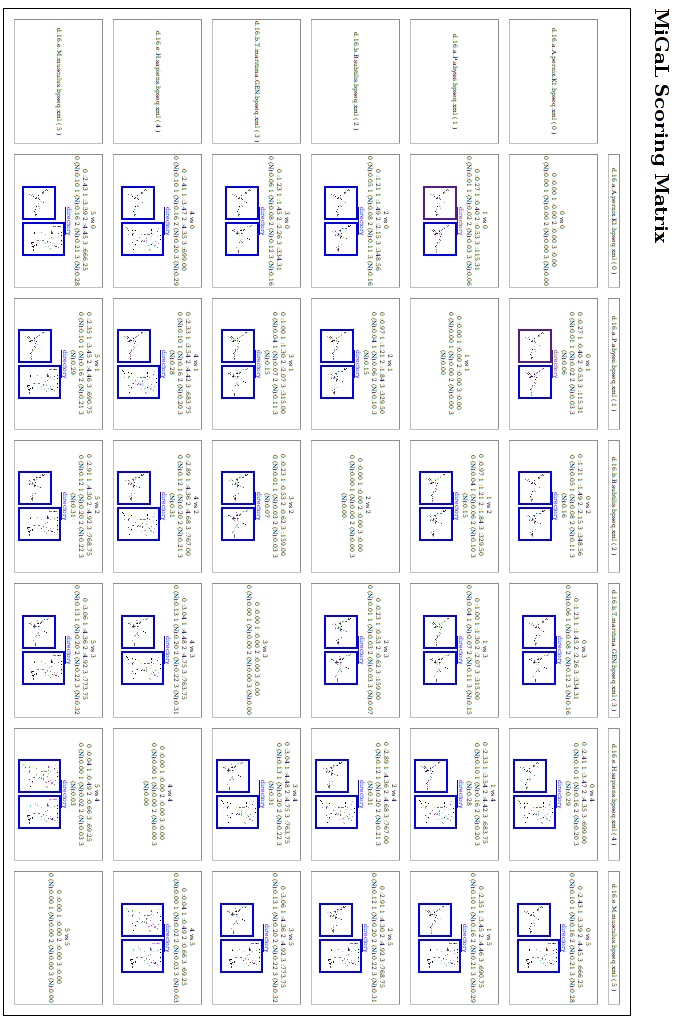
Compute the matrices
To compute the scoring matrices, we use the python
script phylo_nj.py available in the directory tools
of the migal archive. This program runs all comparisons to build the
matrices.
First, we have the problem that structures contains
pseudo-knots.
So, for each comparison we have to tell to migal how to
remove pseudo-knots in the two RNAs involved in the comparison: there
are 15 comparisons!!!
To solve this problem, we have two solutions:
- Convert structures in xml using
rnaconverter. This can be done by using the commandfor i in *bpseq; do rnaconverter -f bpseq $i -i -o $i.xml; done; - Tell
migalto automatically break helices to remove pseudo-knots. This is done by using the option-o "--auto-psdk"withphylo_nj.py.
We suppose that we have converted all bpseq files using rnaconverter
so we have 6 xml files now. Then the command phylo_nj.py *xml
creates 8 matrices in the files Layer0, Layer1,
Layer2, Layer3 and Layer0NORM, Layer1NORM, Layer2NORM, Layer3NORM. Each
file
contains the scoring matrix for one level (0 to 3). The Layer?NORM
corresponds
to the normalized score (see the program description). The file
Layer3NORM should
be:
6
d.16.a.A.p 0.0000 0.0550 0.1636 0.1567 0.2910 0.2765
d.16.a.P.a 0.0550 0.0000 0.1543 0.1473 0.2841 0.2860
d.16.b.B.s 0.1636 0.1543 0.0000 0.0731 0.3141 0.3138
d.16.b.T.m 0.1567 0.1473 0.0731 0.0000 0.3122 0.3153
d.16.e.H.s 0.2910 0.2841 0.3141 0.3122 0.0000 0.0254
d.16.e.M.m 0.2765 0.2860 0.3138 0.3153 0.0254 0.0000
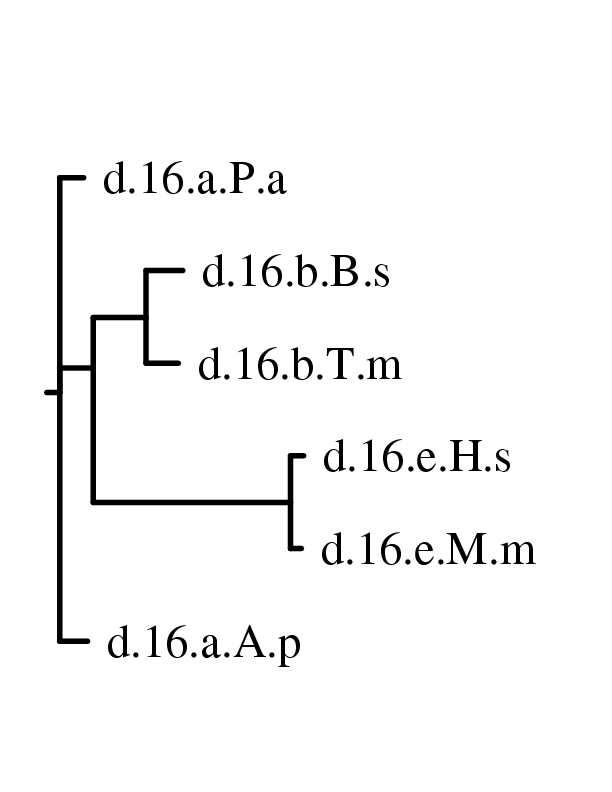
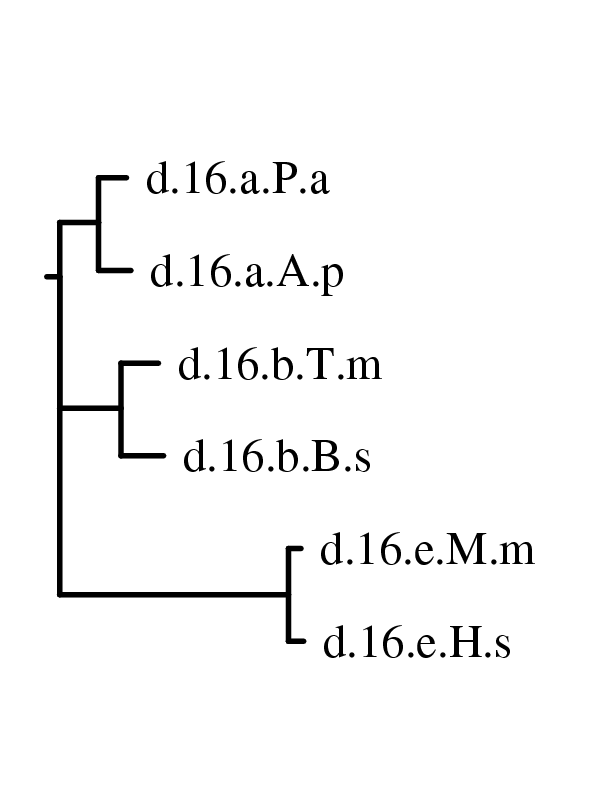
Now one can use these matrices to build a phylogenetic
tree
using the neighbor joining method. If you plans to use phylip
then
phylo_nj can compute the trees. The command phylo_nj.py
--phylip *xml creates 8 postscripts that correspond
to the tree obtained for each matrix by neighbor joining. If one has quicktree
installed, one can ask to compute the tree with quicktree
instead
of phylip with the option --quicktree. The
most significant tree is the one built with Layer3NORM, on the left the
tree built with phylip nj method and on the right the one
built
with quicktree:
 |
 |
Now, if you want to have the alignment resulting
from each comparison, you have to use the option -d.
This option will create a directory for each comparison and run
migal in the directory. Thus the command phylo_nj.py
-d -o "-a align" *xml creates 15 directories. The directory d.16.e.H.sapiens.bpseq_vs_d.16.e.M.musculus.bpseq
contains
this alignement.
Finally, the script phylo_nj.py can also
deal with a postscript file (only format supported by migal).
The option -p indicates to use postscripts if available.
If the postscript files corresponding to
sapiens and musculus are put in the directory and
the command phylo_nj.py -p *xml is run, we obtain
15 directories. In the directories that correspond to a comparison
which involved
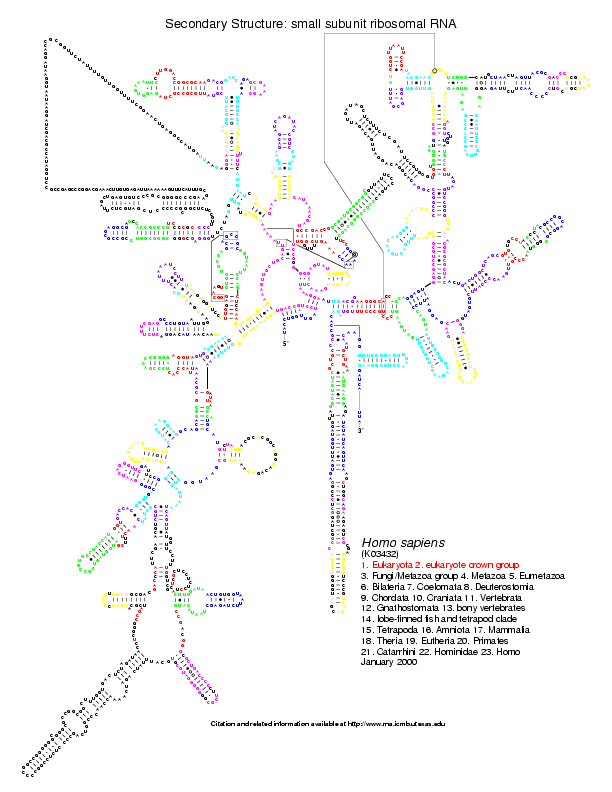
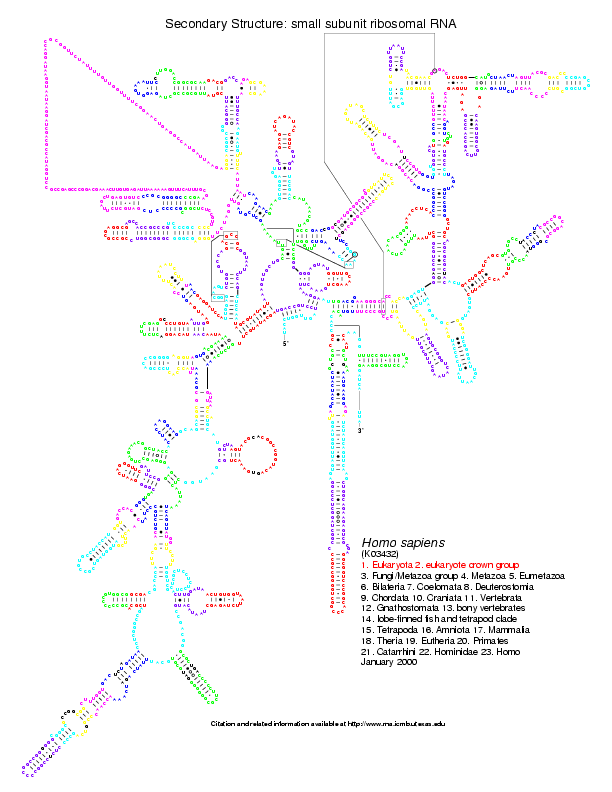
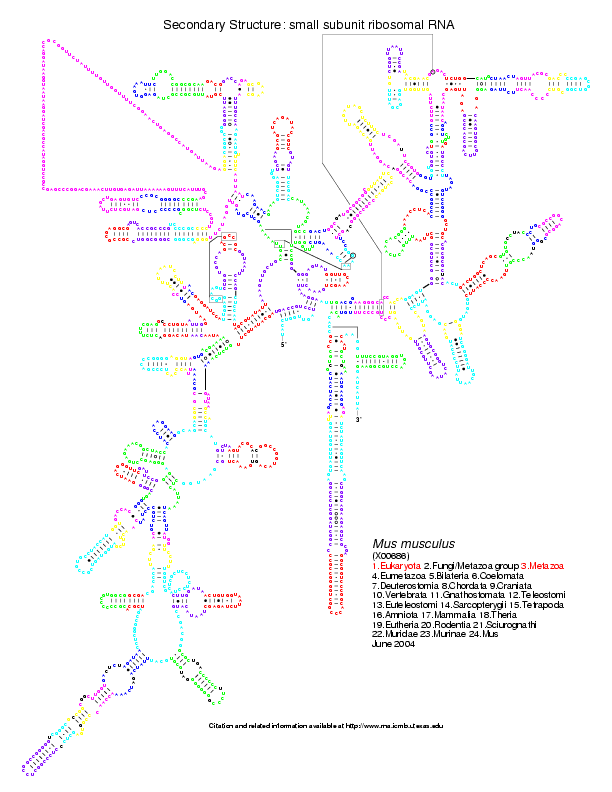
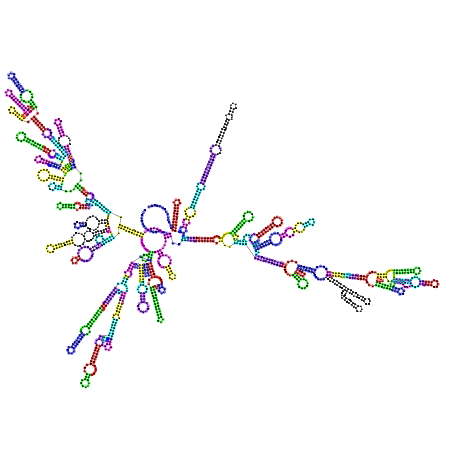
mouse or human, we have the coloured version of the postscripts. Below
is the Homo sapiens 16S coloured from the comparison with Aeropyrum
pernix (on the left)
and Bacillus subtilis (on the right):
|
|
|
|
--rnaplot which tells to phylo_nj.py
to compute missing postscripts with RNAplot(Vienna
package). The command phylo_nj.py --rnaplot *xml builds
four postscripts (for pernix, abyssi, subtilis and maritima) and all
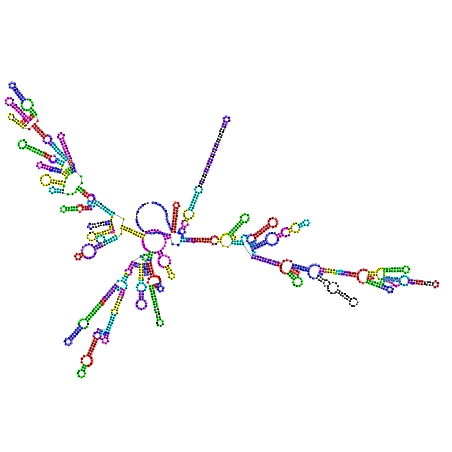
directories contain two postscripts. Below the files in d.16.a.P.abyssi.bpseq_vs_d.16.b.T.maritima.GEN.bpseq:
|
|
Creating an HTML table
The options --html-rel and --html-abs
allow to create an html file that contains a summary of the job run
(include
picture, links to directory, alignments...). In our example, the
command phylo_nj.py --html-rel --rnaplot -o "-a align" *xml
produces this page.